The focus of our research is toward the application of a novel epigenetic profiling assay for transposase-accessible chromatin with high throughput sequencing (ATAC-seq) for integrative epigenomic analysis (Buenrostro et al., 2013). ATAC-seq is a new protocol to capture open chromatin sites by performing adaptor ligation and fragmentation of open chromatin regions. Due to its efficiency in requirement of biological sample and in library preparation time, many scientists are generating ATAC-seq libraries to decipher the chromatin landscape of DNA in a given cell type and condition of interest.
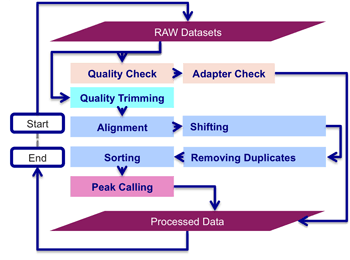 ATAC-seq data processing pipeline’s workflow starts with the quality check and adapter trimming, then alignment, shifting, removing duplicates, sorting and peak calling is performed to find significant numbers of mapped reads, indicating the presence of gene regulatory regions. Implementation of ATAC-seq data processing pipeline is a complex task for biologist (without programming skills), as it involves the I/O (input/output) redirectional integration of several non-interactive command-line applications in Unix, Linux and DOS environments.
ATAC-seq data processing pipeline’s workflow starts with the quality check and adapter trimming, then alignment, shifting, removing duplicates, sorting and peak calling is performed to find significant numbers of mapped reads, indicating the presence of gene regulatory regions. Implementation of ATAC-seq data processing pipeline is a complex task for biologist (without programming skills), as it involves the I/O (input/output) redirectional integration of several non-interactive command-line applications in Unix, Linux and DOS environments.
Here, we present I-ATAC (Interfacing Assay for Transposase-Accessible Chromatin with high-throughput Sequencing) as the interactive, user-friendly, cross platform and open source desktop application, which supports transparent, reproducible and automatic generation of ATAC-seq data quality check and pre-processing.
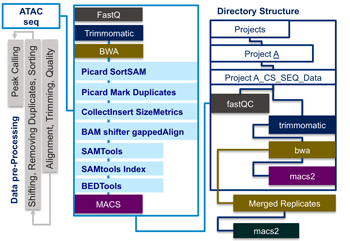 I-ATAC integrates several non-interactive mode applications for quality checking, adapter trimming, alignment, shifting, removing duplicates, sorting and peak calling by automatically generating and running sequential, multiple-parallel and customized data analysis pipelines.
I-ATAC integrates several non-interactive mode applications for quality checking, adapter trimming, alignment, shifting, removing duplicates, sorting and peak calling by automatically generating and running sequential, multiple-parallel and customized data analysis pipelines.
Its performance has been tested and delivered direct results on a private and public large-scale datasets which can be visualized using available genome browsers, for differential expression analysis to find significant numbers of mapped reads, indicating the presence of gene regulatory region in the genome.
I-ATAC following features:
- Connects to the data cluster.
- Locates sample data files.
- Manipulates compressed files.
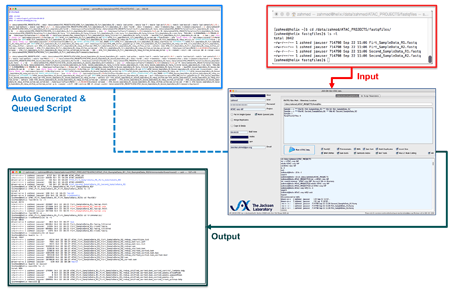 Writes Linux commands.
Writes Linux commands. - Creates directory structure.
- Loads compilers & interpreters.
- Calls integrated applications.
- Creates shell scripts.
- Customizes pipelines.
- Sets processing parameters.
- Sets wall time.
- Sets status notifications.
- Submits and queues jobs.
- Processes data files.
- Performs file management.
- Disconnects data cluster.
The targeted end users of I-ATAC are mainly the biologists, who are used to of interactive operating systems (e.g. Windows, Mac-OS-X) and have no programming experience in command-line environment. I-ATAC is a user-friendly desktop application, based on the fundamental software engineering principles and human computer interaction guidelines. It is programmed in Java and requires Java Runtime Environment to be installed on in-use operating system (e.g. Windows, MacOSX etc.). Additionally, it compels all integrated applications to be downloaded and installed in data cluster and referenced genome for mapping.
Download I-ATAC
I-ATAC is very easy to install, cross platform application, freely available to download and use.
Download - I-ATAC for MAC OS X
Download - I-ATAC FOR WINDOWS OS
To learn more about I-ATAC please contact:
Asst. Prof. Duygu Ucar, Ph.D.
Email: duygu.ucar@jax.org






