The Howell Lab
Investigates the early stages of age-related neurodegenerative diseases.
Principal Investigator
Location
Topics
Our Research Focus
We apply genetics and genomics approaches to identify fundamental processes involved in the initiation and early propagation of age-related neurodegenerative diseases, focusing on Alzheimer's disease, non-Alzheimer's dementia and glaucoma. Understanding these processes provides the greatest opportunity of therapeutic intervention. Another aim is to combine knowledge from human genetic studies with the strengths of mouse genetics to develop new and improved mouse models for dementias and make them readily available to the scientific community.
Full Scientific Report
Identifying critical early events in Alzheimer's disease (AD)
Current mouse models develop AD-related phenotypes such as plaque deposition and glial activation, but the key early events in the pathogenesis of AD have not been studied. Identifying these early processes will provide novel targets to evaluate for human treatments (Figure 1). We are performing gene expression profiling combined with bioinformatics analyses and subsequent functional tests to determine early molecular processes of AD. I first applied this experimental design to glaucoma, where early pathological events were identified and their importance then tested using both gene knockouts and pharmacological intervention.
Bioinformatics approaches are being used to determine genes and biological processes that change early. Gene profiling datasets of this complexity do not currently exist for AD and will greatly facilitate our understanding of molecular changes that occur prior to its traditionally recognized hallmarks. This work is also likely to order processes that are currently known to be involved in AD but whose timing of activation is unclear. Our long-term goal is to determine the beneficial or damaging role(s) that key early events play using genetically engineered mice.

Figure 1: Amyloid-beta plaques first appear in B6.APBTg mice between four and six months of age.To determine the ideal ages at which to perform gene expression profiling, we used the timeframe of AD phenotype development in the cortex. Plaques were visualized using Thioflavin T (seen in blue). The earliest and smallest plaques appear at four months of age.
Developing the next generation of more predictive mouse models for Alzheimer’s disease
Standardizing and improving existing mouse models of AD: Animal models of AD have provided important insights into the pathophysiology of disease and suggested potential avenues for therapies. However, translation of these findings to the clinic has been limited. Therefore, to improve existing models, we are performing standardized behavioral testing on the most popular AD models in collaboration with Genetic Resource Sciences at JAX. We are generating APP/PS1 mice on at least eight genetically diverse strain background to assess whether different genetic backgrounds can improve AD-relevant phenotypes (Figure 2).

Figure 2: Phosphorylated Tau is present in D2.APBTg mice. Hyperphosphorylated Tau protein accumulates in neurons. This accumulation is a hallmark phenotype of AD in humans but is rarer in mouse models for AD that overexpress mutant APP and PSEN1.
Validating genome-wide association hits: Variations in multiple genes are likely to interact to cause late-onset Alzheimer’s disease (LOAD). Hundreds of genes have been associated with LOAD, with meta-analysis significantly associating dozens of them. However, for most genes, definitive role(s) in Alzheimer’s disease have not been identified. Endocytosis is consistently implicated as a key process in AD. APOE, CLU, BIN1, ABCA7, PICALM, CD2AP and SORL1 can be directly linked to endocytosis with additional genes indirectly linked. However, these genes are expressed in multiple cell types and so it is not clear whether disruption to endocytosis in only one cell type, such as in neurons to regulate synaptic function, or in multiple cell types, such as astrocytes and microglia, is a key factor in Alzheimer’s disease. Given that some of the genes involved in endocytosis also function in other processes, it is also possible that variations in these genes impact other aspects of AD, including neuroinflammation and vascular dysfunction.
Identifying novel genes/causative variants for AD: We are working with other investigators at JAX—Dr. Greg Carter, Dr. Michael Stitzel and Dr. Duygu Ucar—to analyze publicly available human genome sequences relevant to AD, including the Alzheimer’s disease sequencing project (ADSP) and Alzheimer’s disease neuroimaging initiative (ADNI), to identify novel variants and loci associated with AD. These putative variants are being engineered in mice using genome editing (CRISPR/cas9) and cohorts of mice aged and phenotyped. We are particularly interested in modeling the heterogeneity of AD by assessing the impact of multiple variants.
Understanding the impact of lifestyle choices in aging and dementia
Exercise improves loss of neurovascular unit decline seen in age
Interestingly, NVU decline was not reversed in APOE deficient mice that already show vascular comprise. This suggests APOE is either a key component of exercise-mediated neurovascular unit preservation or that exercise can prevent but not reverse vascular compromise. Studies are ongoing to determine the factors involved in age-related vascular compromise, the mechanisms by which exercise prevents vascular compromise and the role of APOE in these processes.

Figure 3: Pericytes important to the neurovascular unit lose integrity with age, leading to a breakdown of the blood-brain barrier. Transmission Electron Microscopy shows degeneration of pericytes in aged neurovascular unit (right) as well as thickening of the basement membrane. These pathologies indicate a disfunction in the neurovascular unit, which leads to blood-brain barrier dysfunction. BV = Blood vessel. EC=Endothelial cells. P=Pericyte. DP=Degenerating pericyte.
We are particularly interested in the role of neuroinflammation in aging. We, and others, have shown the complement cascade is upregulated in myeloid cells in aging, and we hypothesize that it may contribute to both neuronal dysfunction (particularly synapse dysfunction) and vascular compromise in aging and very early stages of AD.
A western diet induces chronic neuroinflammation and cognitive deficits
Today, one in three adults in America are obese, a sharp increase since 1950, when only 9% of Americans were obese. Mid-life obesity increases the risk for Alzheimer’s disease and cognitive decline by six-fold, and 20% of Alzheimer’s cases in the U.S. are caused by physical inactivity. It is unknown how the increase in the number of older, obese adults will affect the healthcare system as they age.
Obesity increases the risk of cognitive decline and dementias, but the mechanisms involved are not known. Our data suggests neuroinflammation (inflammation in glial cells such as astrocytes and microglia) is a critical component of obesity-induced cognitive decline. In the brain, astrocytes and microglia are rapid first responders to insults, such as inflammatory cytokines and chemokines. Both astrocytes and microglia are extremely important to brain health, providing nutrients and removing waste from neurons as well as immune surveillance, but they have adverse effects if activated over a sustained period of time (chronic inflammation). Microglia are the brain’s resident macrophage, providing crucial immune responses.
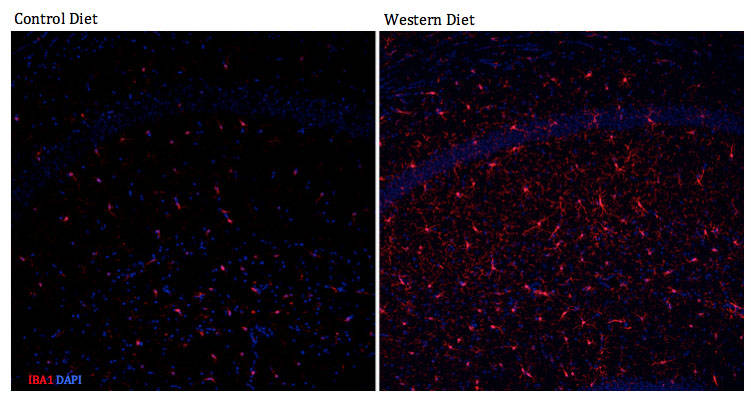
Figure 4: Increased reactive microglia in the hippocampus of western diet-fed C57BL6/J mice. The neuroinflammatory response to a western diet (WD) can be detrimental to the health of neurons, which can lead to cognitive impairment. Microglia become reactive in response to inflammation and remove waste. Once neuroinflammation becomes chronic, microglial response could be detrimental.
Excessive neuroinflammation can influence communication between the brain and the peripheral immune system during disease states. In some neurodegenerative diseases, including AD, peripheral immune cells, such as monocytes, cross the blood-brain barrier to respond to inflammation. This infiltration of monocytes from the periphery has been shown to cause more damage. Resident microglia can also become chronically inflamed or reactive (Figure 4), which can lead to neuronal impairments, including synapse and myelin loss. We are working to better understand obesity-induced neuroinflammation and peripheral immune cell infiltration into the brain (Figure 5), as well as ways to ameliorate inflammation by exercising or by genetically disrupting monocyte infiltration.
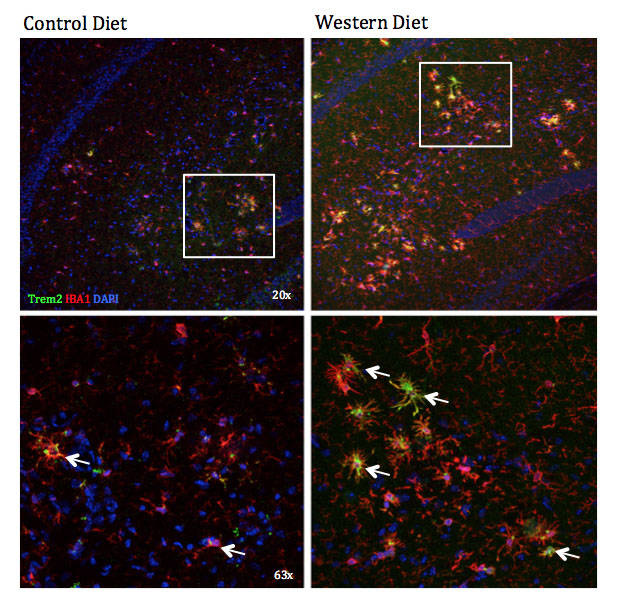
Figure 5: Increased reactive monocytes in response to western diet (WD) in B6.APBTg mice. The neuroinflammatory response to a western diet can be detrimental to the health of neurons, which can lead to a cognitive impairment. The number of microglia (labeled by IBA1 in red) is increasing in response to WD in the hippocampus. This is likely due to peripheral immune cells infiltrating into the brain in response to inflammatory cytokines. Trem2 (green) is expressed on monocytes/macrophages (including microglia) in environments of severe inflammation, including in neurodegenerative diseases like AD.
Investigating cell specific roles of complement cascade in glaucoma
In addition to our interests in the role of the complement cascade in aging and dementias, we are studying its role in glaucoma, a disease that affects 70 million people. Glaucoma is a leading cause of blindness worldwide, characterized by the loss of retinal ganglion cells (RGCs), the output neurons of the retina. Age and intraocular pressure elevation are major risk factors for glaucoma. Complement expression increases in human glaucoma and in all animal models of glaucoma assessed to date. Our work extended previous studies by showing that induction of the complement cascade occurs very early during glaucoma (in both RGCs and monocyte-derived cells) (Figure 6). We found that DBA/2J mice with a targeted null mutation in the gene for complement component 1qa (C1qa) are robustly protected from glaucoma. The robust protection highlights the importance of determining exactly how the complement cascade influences glaucoma. C1QA is expressed in at least two cell types (RGCs and microglia). Complement component C3 is also expressed in astrocytes. We are functionally testing the importance of C1Q and C3 from neurons, myeloid-derived cells and astrocytes using conditional knockout mice. Determining the significance of complement biosynthesis by each of these cell types is important for designing therapeutic interventions that manipulate the complement system.
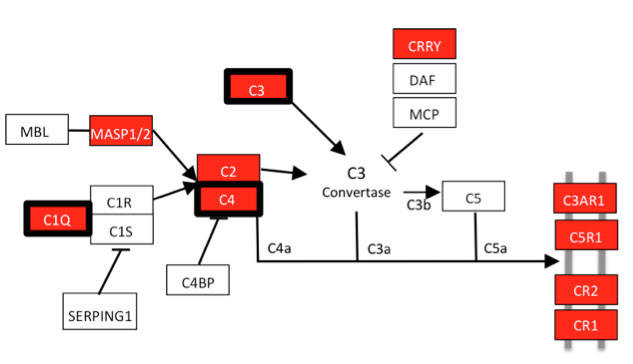
Figure 6: Molecular clustering identifies early stages of glaucoma in the retina. Pathway analysis (using DAVID) of differentially expressed genes in two early stages of glaucoma identified the complement cascade as the most significantly over-enriched pathway. Genes shown in red have greater expression in the second molecularly defined retinal stage. The three genes marked by red and bold boxes were differentially expressed in the earliest retinal stage.
-
 Media ReleaseOctober 09, 2024
Media ReleaseOctober 09, 2024Study probes how eating less can extend lifespan
Researchers at The Jackson Laboratory conduct pivotal study into aging and lifespan to uncover new details about how diets might make people live longer — but also their negative side effects -
 ArticleJuly 24, 2024
ArticleJuly 24, 2024Researchers are closing in on a mouse model for late-onset Alzheimer’s research
Mice don’t get Alzheimer’s—and while that’s good news for mice, it’s a big problem for biomedical researchers seeking to understand the disease and test new treatments. Now, researchers at The Jackson Laboratory are working to create the first strain of mice that’s genetically susceptible to late-onset Alzheimer’s, with potentially transformative implications for dementia research. -
 Research HighlightJuly 10, 2024
Research HighlightJuly 10, 2024Charles Lee continues to sequence repetitive areas of the genome
Charles Lee co-leads the Human Genome Structural Variation Consortium and is uncovering structural variations in the human genome. -
 Featured ArticleJuly 09, 2024
Featured ArticleJuly 09, 2024What is genetic biomarker testing?
Genetic testing is a powerful tool in the fight against cancer, playing a crucial role in both prevention and personalized treatment. But what exactly are the different types of genetic tests, and how do they help?
-
 ArticleJuly 29, 2024
ArticleJuly 29, 2024MODEL-AD drives progress for Alzheimer’s research
The July issue of Alzheimer’s & Dementia spotlights progress by MODEL-AD and its work to create better models for Alzheimer’s disease research and catalyze progress in the field. -
 Media ReleaseJuly 24, 2024
Media ReleaseJuly 24, 2024New analysis - targeted Alzheimer’s research and drug discovery
Researchers at The Jackson Laboratory offer the first comprehensive ranking of the relative role and significance of every known gene and protein in the development of Alzheimer’s Disease in advance of the Alzheimer's Association International Conference, July 28 -
 Featured ArticleJuly 09, 2024
Featured ArticleJuly 09, 2024Bringing critical perspectives to genomic medicine research
Leading with compassion, Elizabeth Charnysh represents a team of genetic counselors established seven years ago as part of a comprehensive strategy to study the impact of genomic medicine research on human health. -
 Blog PostJune 27, 2024
Blog PostJune 27, 20245 things: research tools and methods that may be medical game changers
Explore five research tools and methods that didn't exist ten years ago that could be medical game changers.