New methods allow researchers to figure out what happens where in tumors, with important implications for improving cancer therapies.
“Cancer is complex.” That statement has always been true, but it has meant different things over the years.
About 100 years after scientists first determined that tumors are composed of the body’s own over-proliferative cells, the World Health Organization’s Classification of Tumours was published in a series of book volumes. The tomes expanded on the standard classification by tissue or organ of origin, subtype, grade according to WHO and spread (metastasis). Nearly 50 years ago, genetic data started to come into play with the discovery and characterization of oncogenes such as MYC and HER2, as well as the consequences of mutations in genes such as BRCA1/2 and TP53.
Fast forward to a decade ago and the promise of precision oncology was looming large. The development and implementation of next-generation genome-sequencing technologies meant that, at least in theory, both tumor and normal tissue could be sequenced, the molecular causes identified and targeted therapies applied. It represented an important step forward from classifying cancers mostly according to site of origin, and from the toxic chemotherapy and radiation treatment regimens that kill all dividing cells. Cancer’s complexity could actually be characterized and, perhaps, used against it in precise ways. It was an exciting thought.
It was in this context that a sobering paper was published in Cancer Medicine. Researchers had done a deep dive into kidney cancer and found that crucial differences existed in biopsy samples obtained from different areas of the same tumor. They even recommended that three to five sites be biopsied from each tumor before treatment decisions were made. I distinctly remember reading the paper and associated commentaries when they came out and feeling deflated. Just as we seemed to be getting one step ahead of cancer, its biological realities brought us back to earth. If one tumor harbored enough variability to thwart precision therapies, what chance did we have of advancing cancer care?
Deconvoluting cancer
Of course, the decade since has seen exciting progress on many fronts, especially the emergence of cancer immunotherapies. But genome sequencing and targeted therapies have remained somewhat hit and miss, in part because of the intra-tumor variability, called heterogeneity, previously identified, as well as the development of treatment resistance mechanisms in many cancers. Researchers have therefore moved well past the “multiple biopsy site” paradigm to investigate cancers and their immediate environments, and they can now do so with astonishing resolution. A new language is emerging around these efforts, and a key phrase at the moment is “deconvoluting spatial transcriptomics.” But what does that mean and why is it important for cancer?
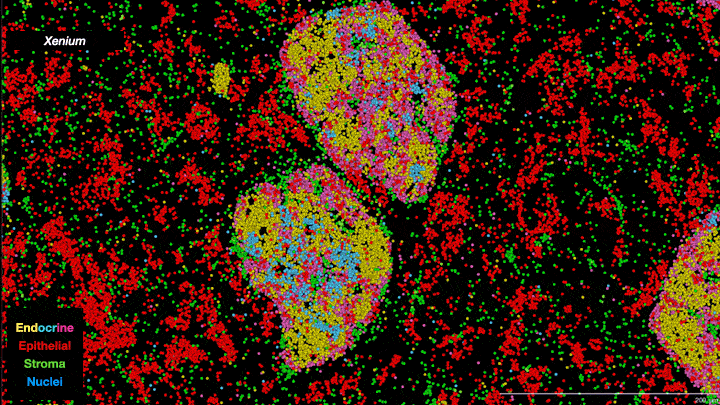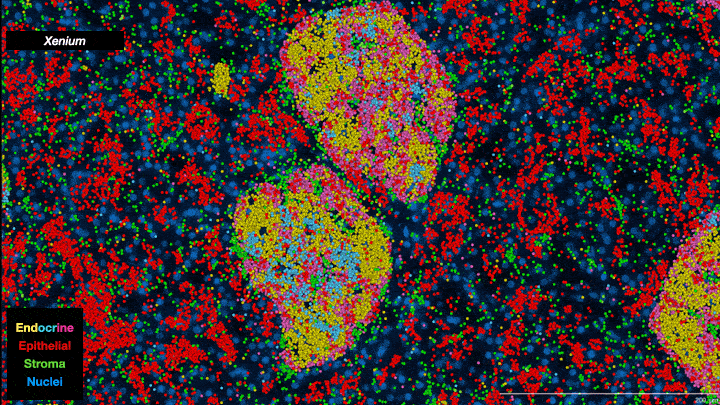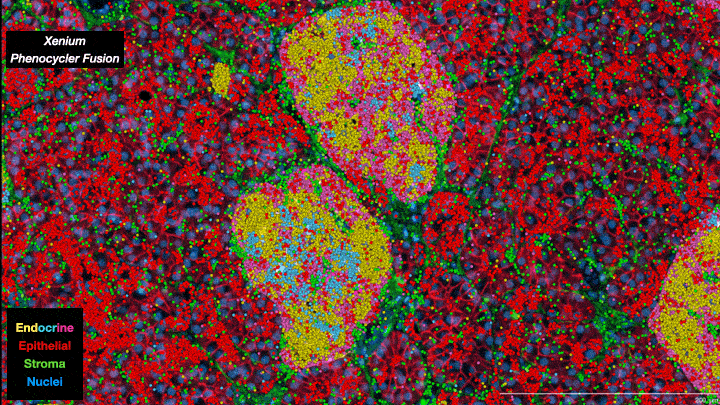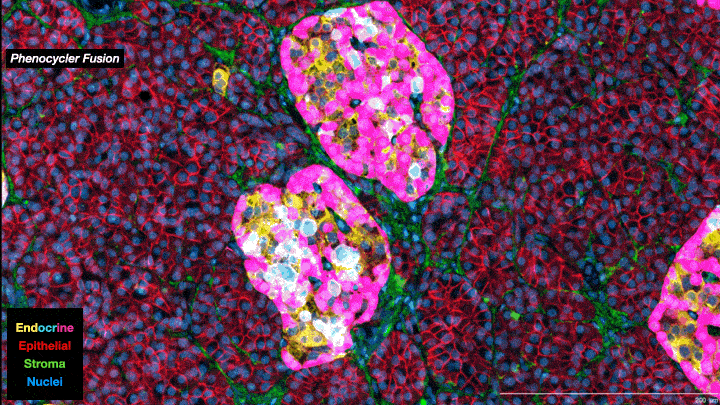
In essence, scientists are now able to analyze the full collection of messenger RNAs (mRNAs) present in individual cells on a cell-by-cell basis. Known as the transcriptome, it provides information about which genes are being expressed, how active they are and what proteins are likely to be active in the cell. It’s not a direct relationship—actual protein abundance is determined by multiple factors—but because mRNAs are the primary intermediaries between gene expression and protein production, they provide important insights into cell types and function. Single-cell transcriptomes are allowing researchers to characterize what cells are located where within tumors, including different subtypes of tumor cells as well as “normal” cells associated with the tumor such as immune cells and structural cells. And when combined with other ’omics data—genomics, proteomics, metabolomics, epigenomics—the information obtained provides a truly unprecedented understanding of cellular mechanisms and cancer biology. As one review paper describes it, it is a journey “into the cancer multiverse.”
Can we stay ahead of cancers?
While all of the biological insight is valuable in and of itself, the desired goal for acquiring all the data is, of course, finding better ways to treat cancers. The more we know about cancer biology, the more we know about exactly how it begins, grows, evolves, resists treatment and more. That knowledge also holds the key to finding possible weaknesses and dependencies, providing new molecular pathways and mechanisms to target therapeutically. And The Jackson Laboratory (JAX) is driving progress in the field. Professor Jeffrey Chuang, Ph.D., began analyzing tumor evolution and heterogeneity using pathology images several years ago, but he predicted the implementation of spatial transcriptomics and its implications ahead of time. He and other researchers also work closely with the JAX Single Cell Biology Laboratory, which has contributed to many fields in addition to cancer—the pathology of endometriosis, the biology of senescent cells and the discovery of neuronal subtypes come to mind—but has also provided key insights into tumor subtypes, tumor composition and heterogeneity. Recent progress, made in a collaboration with Professor Paul Robson, Ph.D., and the SenNet consortium, includes the development of a new protocol for visualizing both mRNA transcripts and proteins within the same tissue section. Senescent cells are thought to contribute to diseases of aging, including cancer, and SenNet is seeking to better characterize their biology and how they contribute to disease.
The research advances have yet to be fully implemented in the clinic, but even today they are having an effect. The Maine Cancer Genomics Initiative (MCGI), a program designed to accelerate the use of precision oncology and genome-matched treatments in the clinic, is at the forefront of using tumor ’omics data to guide treatment. Emerging findings indicate that patients see important benefits from current targeted therapies, underscoring the importance of additional progress. At the recent MCGI Forum, the keynote speaker, Razelle Kurzrock, M.D., from the Medical College of Wisconsin, presented her work with “N-of-1” therapeutic strategies, in which each patient is thoroughly tested and treated as a unique case. Obtaining the large amounts of tumor and normal tissue data, as well as devising unique combination treatment strategies, remains challenging, but the early results are impressive. With the right resources and expertise, knowing more about each tumor clearly leads to better patient outcomes.
Ultimately, “deconvoluting” each patient’s tumor—being able to determine exactly what is going on where within the tumor—promises to be vital for figuring out how to attack cancer, overcome therapy resistance and prevent recurrence. Cancer’s complexity remains daunting, but we are entering a new era of understanding it and, hopefully, delivering even better care.